Metrics
Message boards : Number crunching : Metrics
| Author | Message |
|---|---|
|
Mark Send message Joined: 10 Nov 13 Posts: 40 Credit: 397,847 RAC: 0 |
Hi, I was interested in the metrics/efficiency of the search as a research area. My understanding is that Rosetta@Home randomly picks a start point then uses simulated annealing to find a minima. Multiple decoys are produced from multiple computers and plotted on a graph such as this. (In this example it shows a nice funnel with the folds of interest towards the turquoise area in bottom left)  My question is how many data points are generated per protein investigation and how much CPU time is required to produce one point? Obviously larger proteins with more residues are going to take longer/more effort. Is there a rough relationship between number of AA to search effort? Thx Mark |
|
Mod.Sense Volunteer moderator Send message Joined: 22 Aug 06 Posts: 4018 Credit: 0 RAC: 0 |
Some proteins are studied in phases, where results from one phase are used to place some constraints on the search space of subsequent phases. So I'm sure there is no simple answer to your question. To gain an intuitive sense of it, you might spend some time playing FoldIt! I believe the total potential search space is on the order of the number of AAs cubed. Even that depends upon the specific AAs. But the search algorithms are able to do better than that. And it's a good thing, otherwise you'd need billions of years of all existing computing power to perform an exhaustive search on a large protein. Rosetta Moderator: Mod.Sense |
|
Mark Send message Joined: 10 Nov 13 Posts: 40 Credit: 397,847 RAC: 0 |
Some proteins are studied in phases, where results from one phase are used to place some constraints on the search space of subsequent phases. So I'm sure there is no simple answer to your question. Thx for that. I have tried foldit and do have a sense of the difficulty. Does anyone have any actual hard data though on the amount of compute required for a particular protein? |
 David E K David E KVolunteer moderator Project administrator Project developer Project scientist Send message Joined: 1 Jul 05 Posts: 1480 Credit: 4,334,829 RAC: 0 |
The "Sampling bottlenecks in de novo protein structure prediction" paper provides some insight. The quick answer is that it depends on the protein. |
|
Mark Send message Joined: 10 Nov 13 Posts: 40 Credit: 397,847 RAC: 0 |
The "Sampling bottlenecks in de novo protein structure prediction" paper provides some insight. OK thx for that. I see it gives a rough figure of say 1000-10000 samples to get an acceptable RMSD (for an approx 100 residue protein)[Fig 3], and how using specific constraints cuts that down. However the paper is about 6 years old and obviously a) Rosetta has moved on since then and b) computing speed has moved on since then through Moores law. Also the paper didn't mention (that I noticed) actual time taken for each of those samples to finish, obviously a factor. I take the point the analysis is done in stages too. However I still don't know how many compute hours to get to a 2Å RMSD decoy for an "average" 100 residue protein...? Is much performance testing done? I would think someone has the Rosetta@Home figures for how many decoys are sent out for a given protein and how long they take? Is the information sensitive in some way? Thx |
|
David E K Volunteer moderator Project administrator Project developer Project scientist Send message Joined: 1 Jul 05 Posts: 1480 Credit: 4,334,829 RAC: 0 |
Definitely not sensitive info :) The algorithm for ab initio folding has not changed much since. Our Robetta automated structure prediction server which uses R@h for sampling aims for around 20,000 decoys of the target and 5,000 decoys from up to 4 homologs. Assuming a runtime of around 2 minutes per decoy for a 100 residue protein gives around 56 cpu days for a somewhat modern cpu. Of course there are many factors that determine the runtimes and they vary depending on the specific types of jobs and targets being run, and of course the computer. I'll see if anyone in our lab can contribute to this discussion. I'm pretty sure someone doing protein design and forward folding (ab initio tests on the designed sequences) can chime in. But keep in mind, experiments may vary considerably in their computing requirements etc.. Runtimes and decoy counts are given for each job in the stderr output which is available for each job you run. If you look at the tasks from your computer(s) you can click on the Task ID to get this info. |
|
TJ Volunteer moderator Project developer Project scientist Send message Joined: 22 Oct 10 Posts: 9 Credit: 216,670 RAC: 0 |
Definitely not sensitive info :) David's estimates are spot on. & I'll second that the that times & number of decoys is protein dependent. Some proteins fold great with 100 decoys while others require > 100k decoys. There's certain things about a protein topology that makes it very difficult to solve (for example long loops). Proteins with difficult topologies take a lot more time to solve. For protein design problems we choose simple topologies which we can fold with <1000 decoys. That's why we can test lots of designs in a limited amount of time. |
|
Mark Send message Joined: 10 Nov 13 Posts: 40 Credit: 397,847 RAC: 0 |
Definitely not sensitive info :) Ah brilliant, that's the kind of thing I was looking for! So 56 cpu days is a rough ballpark for a 100 residue protein (with wide variation!) :) Thx also to TJ. Yes I appreciate there is a large range of "difficulty of solving", maybe I should have used the term "median" rather than "average"! Does anyone happen to have say 3 test proteins with solving stats that I could play with? I am currently using a protein that was sent out to Rosetta@Home some time in Oct (fasta:ACSGRGSRCPPQCCMGLRCGRGNPQKCIGAHEDV) which I think I pulled out of the software correctly! To get some solving stats for it would be great. Looking at the stats you mention, the cpu time is the time for that job on my PC, although the same job (with different seeds presumably) will have been sent to other PCs as well? Is there any way to see them (the job name is not hyperlinked) and hence give the the total time (over all PCs) for that protein? Also I note the stderr doesn't give the residue count so unless I grab each protein myself, I won't have that data either.... Thx to all for pointing me in the right direction! Mark |
|
Timo Send message Joined: 9 Jan 12 Posts: 185 Credit: 45,661,974 RAC: 0 |
Definitely not sensitive info :) I started looking at a couple of my tasks and most of them say they've completed between 6 and 10 decoys (An example here) - what am I missing because that would work out to 40-60 minutes per decoy considering the total CPU time is around ~6 hours?? I am as curious as Mark is about this kind of thing - I work as a Database Developer / Business Intelligence Analyst and one of the things that amazes me/motivates me to contribute to projects like this is that I get frustrated when I have to wait for hours for data crunching queries to run at my day job, I can hardly imagine having to wait 50-60 days for results!!! ... Especially if some of what you're doing is iterative in nature, I try to imagine compiling code/running a query, waiting 60 days for it to crunch, tweaking it a bit and running the tweaked version.. rinse and repeat... that's gotta be really frustrating!?!? |
|
Mark Send message Joined: 10 Nov 13 Posts: 40 Credit: 397,847 RAC: 0 |
Definitely not sensitive info :) I would guess the 56 cpu days is spread over many computers hence they would get results in under 24 hours.... |
|
Mod.Sense Volunteer moderator Send message Joined: 22 Aug 06 Posts: 4018 Credit: 0 RAC: 0 |
Timo, I believe the task you cited is not an ab initio task. There are various types of tasks, in addition to various proteins and potentially "difficult topologies". And then there is protein design as compared to structure prediction of an existing protein. Rosetta Moderator: Mod.Sense |
|
Mod.Sense Volunteer moderator Send message Joined: 22 Aug 06 Posts: 4018 Credit: 0 RAC: 0 |
I would guess the 56 cpu days is spread over many computers hence they would get results in under 24 hours.... Right. The power of distributed computing, right there. No, I'm not aware of any aggregated data on runtimes. As you say, what you can see is just the portion your machine(s) worked on. It is not available by clicking through your tasks. I believe DK was just pointing out that you could look at your own tasks to get a sense of CPU time per decoy... and how it varies. Rosetta Moderator: Mod.Sense |
|
Mark Send message Joined: 10 Nov 13 Posts: 40 Credit: 397,847 RAC: 0 |
No, I'm not aware of any aggregated data on runtimes. See, this is what strikes me as strange. How do you know if new techniques are working OK without measurement? How do you know where bottlenecks and development areas are without measurement? How do I know if an idea I have is better or worse than the current state of play without some kind of metric to measure against? By making performance data available, you would be encouraging new ideas and development of Rosetta as a whole, to the benefit of everyone. The type of thing initially required is so straightforward that it must surely be available somewhere by someone in the R@H program...? What needs to be done to get hold of it? Who do I need to contact? Thx |
|
David E K Volunteer moderator Project administrator Project developer Project scientist Send message Joined: 1 Jul 05 Posts: 1480 Credit: 4,334,829 RAC: 0 |
Rosetta is developed by numerous academic institutions through the Rosetta Commons. There are continuous unit tests, performance benchmarks, etc.. run after each code checkin. The unit and performance tests framework is part of the code distribution. The code is constantly being developed. In fact there's a developers meeting going on right now in Boulder, CO where many developers from the various commons institutions meet to talk about code development, testing, optimization, etc. There's also an annual meeting to talk about the science. If you are talking about a specific protocol benchmark like ab initio folding. We do have benchmark sets that we use for testing etc. |
|
Mark Send message Joined: 10 Nov 13 Posts: 40 Credit: 397,847 RAC: 0 |
Rosetta is developed by numerous academic institutions through the Rosetta Commons. There are continuous unit tests, performance benchmarks, etc.. run after each code checkin. The unit and performance tests framework is part of the code distribution. The code is constantly being developed. In fact there's a developers meeting going on right now in Boulder, CO where many developers from the various commons institutions meet to talk about code development, testing, optimization, etc. There's also an annual meeting to talk about the science. Hi David, yes I'm am focusing on ab initio folding. Are the benchmarks and more specifically, the results available somewhere? I had a quick look through the code distribution and I see the framework to run standardised tests, I see some small test protein cases, but if we are talking 56 days for repeated tests to get to 2Å RMSD, I think I prefer to use your data! :) |
|
David E K Volunteer moderator Project administrator Project developer Project scientist Send message Joined: 1 Jul 05 Posts: 1480 Credit: 4,334,829 RAC: 0 |
Are you looking for decoy sets? If you are looking for runtime benchmarks/data, I believe the decoy sets have runtime info for each decoy, which of course are dependent on the machines that produced them. I can also provide the input files but I can't spend much time helping you with details. |
|
Mark Send message Joined: 10 Nov 13 Posts: 40 Credit: 397,847 RAC: 0 |
Are you looking for decoy sets? If you are looking for runtime benchmarks/data, I believe the decoy sets have runtime info for each decoy, which of course are dependent on the machines that produced them. Hi David, Thx for your reply and your time. Not sure exactly what you mean by decoy sets. Do you mean all the decoys generated for one sequence? Sure that would be interesting, but not essential. I am looking for say 3 protein input/starting sequences of approx 40, 90 and 150 AA length. I presume you start the fold with the residues in a straight long chain? I then just need the total cpu time that R@H contributors spent on it, and number of decoys produced before you got to at least one decoy within 2Å RMSD. (Obviously I could extract the runtime from each decoy and sum if necessary) I'm also unsure of the end criteria R@H uses, where you decide when a sequence is "solved" and you move onto the next? I would guess you estimate the number of decoys required, run that, and then find out if it actually gave a good decoy or not and then maybe repeat if it is an interesting target, or do you do something different? If you could upload those sequences and data somewhere I would be very grateful! Definitely a beer or two for you if you come to London! :) Thx Mark |
|
Mod.Sense Volunteer moderator Send message Joined: 22 Aug 06 Posts: 4018 Credit: 0 RAC: 0 |
Given the conversation that has taken place since I posted this, the OP is probably already aware, but I misspoke there. I should have said that the search space is on the order of 3 to the power of the number of AAs. So 100 AAs is on the order of 5.2 to the 47th. So to exhaustively explore all of those potential conformations would requiring running all of the computers that exist for longer than our solar system has existed... just to explore one protein. Hence, someone's got to come up with a better way. Rosetta Moderator: Mod.Sense |
|
Mark Send message Joined: 10 Nov 13 Posts: 40 Credit: 397,847 RAC: 0 |
Yep thx for that. I knew it was realistically incalculable so I didn't bother to find out by how much! I plotted a graph from the data, and there doesn't seem to be much correlation between protein size and total run duration. This may well be a factor in how the runs are initialised, if a person is estimating how many decoys will be needed for example. Anyway this is what it looks like 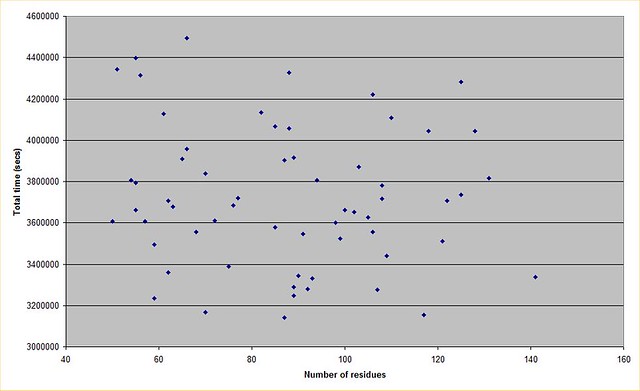 https://farm9.staticflickr.com/8637/16610904862_fb4cd398e2_z.jpg (No idea why the picture is not displaying, tags are correct...?) |
|
Mod.Sense Volunteer moderator Send message Joined: 22 Aug 06 Posts: 4018 Credit: 0 RAC: 0 |
The lack of a trend line for increased decoy count for a larger protein could also be an accounting difference. As was discussed previously in the thread, some proteins are studied in phases and so from start to finish perhaps data for such a protein appears as 4 or 5 separate runs. Rosetta Moderator: Mod.Sense |
Message boards :
Number crunching :
Metrics

©2025 University of Washington
https://www.bakerlab.org